

Setbp1overexpression acts in the place of class-defining somatic mutations to drive mouse and human FLT3-ITD-mutant AMLs
Suruchi Pacharne,1,2 Oliver M. Dovey,1 Jonathan L. Cooper,1 Muxin Gu,1,2 MS Vijaybaskar,1,2 Mathias J. Friedrich,1,5 Malgorzata Gozdecka,1,2 Sandeep S. Rajan,1,4, Etienne De Braekeleer,1,2 Maxim Barenboim,5,6 Grace Collord,1,2 Hannes Ponstingl,1 Ruben Bautista,1 Milena Mazan,1,8 Roland Rad,5,6 Konstantinos Tzelepis,1,7 Penny Wright,3 and George S. Vassiliou1,2,9*
Abstract
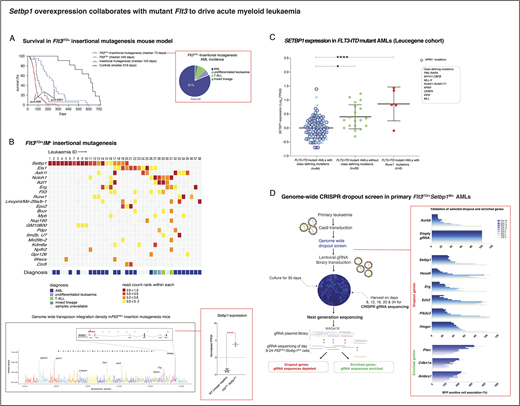
Internal tandem duplications in FLT3 (FLT3-ITD) are found in 30% of acute myeloid leukemia (AML) cases and impart a poor prognosis. FLT3-ITD commonly synergizes with class-defining mutations such as chimeric fusion genes or mutations in NPM1, RUNX1, CEBPA or MLL to drive AML. However, 20% of FLT3-ITD-mutant AMLs bare no class-defining mutations and the mechanisms of acute leukemic transformation in these cases are unknown. To identify pathways that can drive FLT3-ITD-mutant AML in the absence of class-defining mutations, we performed an insertional mutagenesis (IM) screen in Flt3-ITD mice using the Sleeping Beauty transposon system, activated by the Mx1-Cre recombinase in hematopoietic stem cells. All mice developed acute leukemia, predominantly AML, after a median latency of 73 days (Figure A). Analysis of transposon insertions in 38 Flt3-ITD/IM leukemias identified common integration sites (CISs) in 22 loci (Figure B). The most commonly "hit" genes were Setbp1 (20/38), Ets1 (11/38), Ash1l (8/38), Notch1 (8/38), Erg (7/38), Flt3 (6/38) and Runx1 (5/38) (Figure B). Of these, Setbp1 and Runx1 were unique to Flt3-ITD and not identified as CISs in insertional mutagenesis screens of wild type, Npm1c or BCR-ABL-expressing mice. Transposon insertions in Setbp1, primarily located upstream of its first coding exon, were associated with Setbp1 and Hoxa mRNA overexpression and were invariably associated with AML development (Figure B). These findings propose that overexpression of wild type SETBP1 may collaborate with FLT3-ITD to drive leukemogenesis in human AMLs lacking mutations known to collaborate with mutant FLT3. Corroborating this, we found that SETBP1 expression was higher in human FLT3-ITD-mutant AMLs lacking class-defining mutations and in those with RUNX1 mutations (Figure C). We go on to show that Setbp1 insertions activate a Hoxa gene signature such that shares significant similarities, but also specific differences to those driven by mutant Npm1 and MLL fusion genes. We go on to show, using CRISPR-gRNA, that whilst Flt3ITD/+/SETBP1IM+AMLs are entirely dependent on Setbp1 expression, Flt3ITD/+/Npm1cA/+AMLs are not, but do depend on the expression of the homebox gene Nkx2.3. Our findings propose that SETBP1 overexpression activates a gene expression pattern that collaborates with FLT3-ITD to drive many human AMLs and that this combination represents a specific subtype of AML amongst AMLs lacking class-defining mutations. To identify genetic vulnerabilities of this AML subtype, we performed genome-wide CRISPR-Cas9 recessive screens in primary murine Flt3ITD/+SETBP1IM+AMLs and identified more than 2000 genetic vulnerabilities, of which 677 were not required for the survival of HPC7 non-leukemic hematopoietic cells including >100 "druggable" genes such as Brd3, Ezh2 and Hmgcr (Figure D). Collectively our study: i) identifies SETBP1overexpression as a non-genetic alteration driving a subgroup of FLT3-ITD mutant AMLs lacking class-defining somatic mutations and ii) goes on to define the genetic vulnerabilities of such AMLs as a starting point for the development of targeted therapies.
Vassiliou:Kymab Ltd - Monoclonal antibody company. Currently not working in myeloid cancers or clonal haematopoiesis.: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal